Uploaded XML¶
GTG allows uploading an existing XML file and building web components upon it. In this section, we will show how to build
seqtk_seq_2.xml from seqtk_seq_1.xml.
Upload XML¶
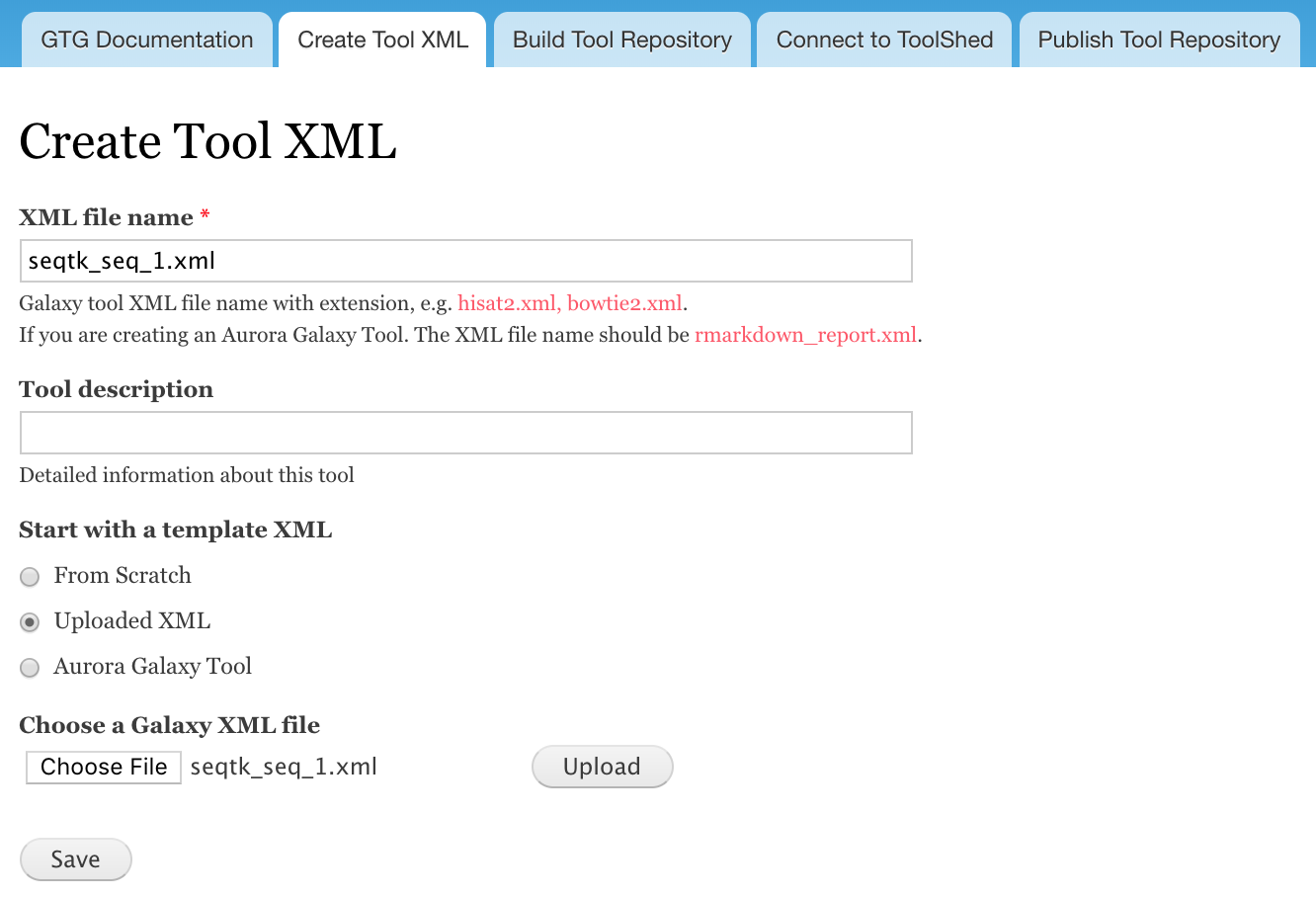
- Click the Create Tool XML tab
- Enter
seqtk_seq_2.xmlinto XML file name - Leave Tool description blank for the tutorial
- Select Uploaded XML
- Click Choose File and select seqtk_seq_1.xml in your computer and click Upload
- Click Save
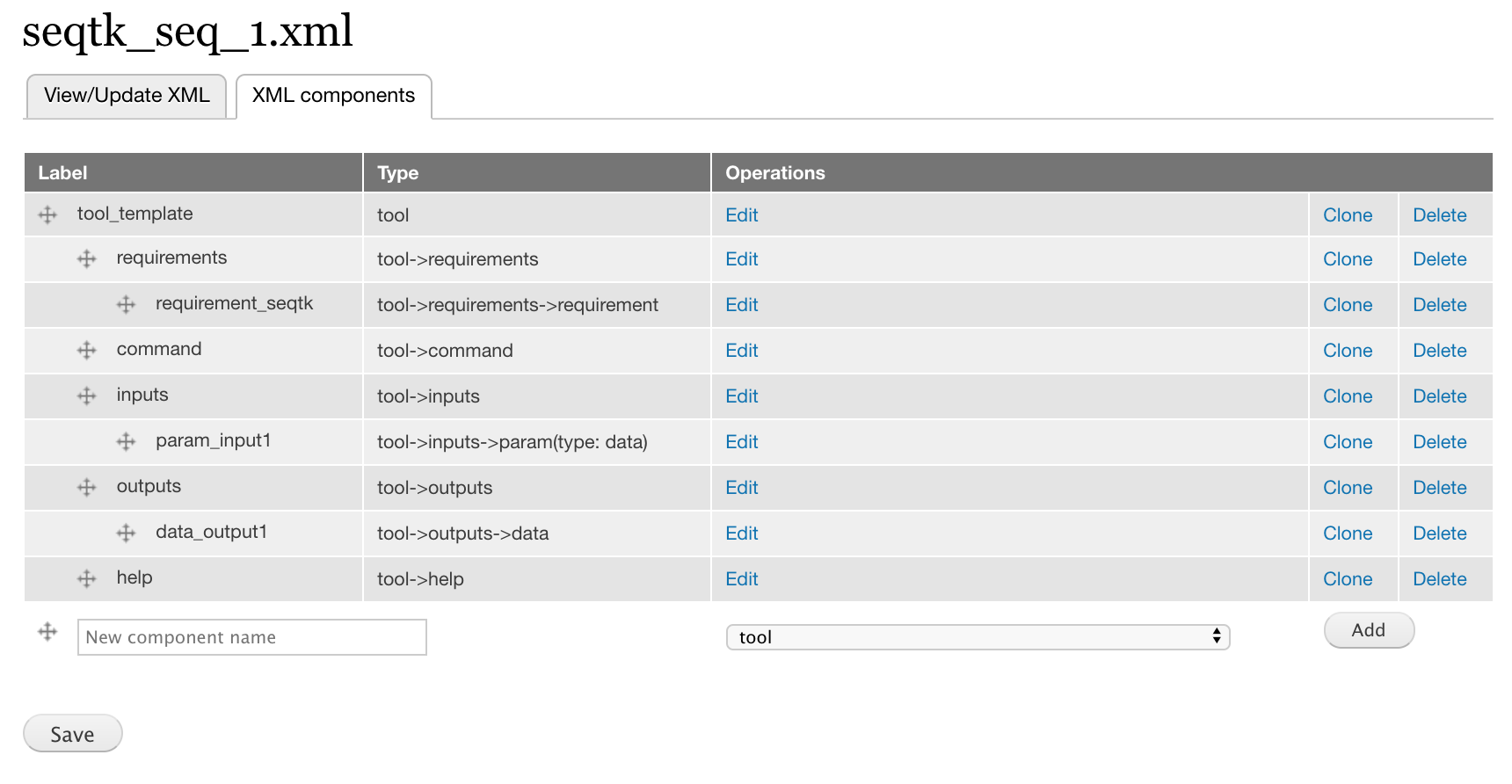
You should be redirected to the webform components page. If not, you can click the Build Tool Repository table, and click edit for the XML you just created.
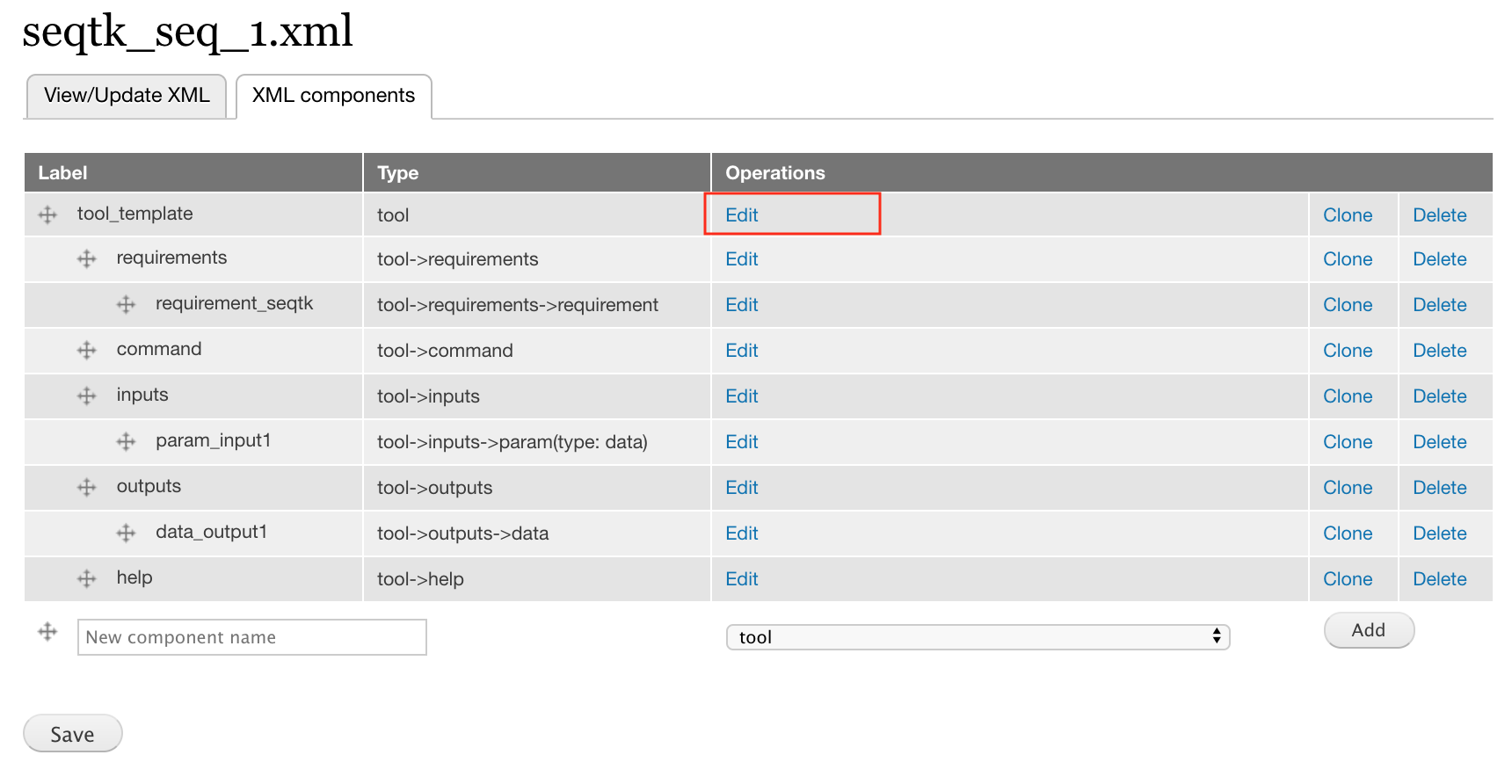
Correct Tool ID attribute¶
When you upload an XML file, the Tool ID attribute in the tool component is always tool_1. We need to correct this attribute.
- Click edit for the tool component on the component page.
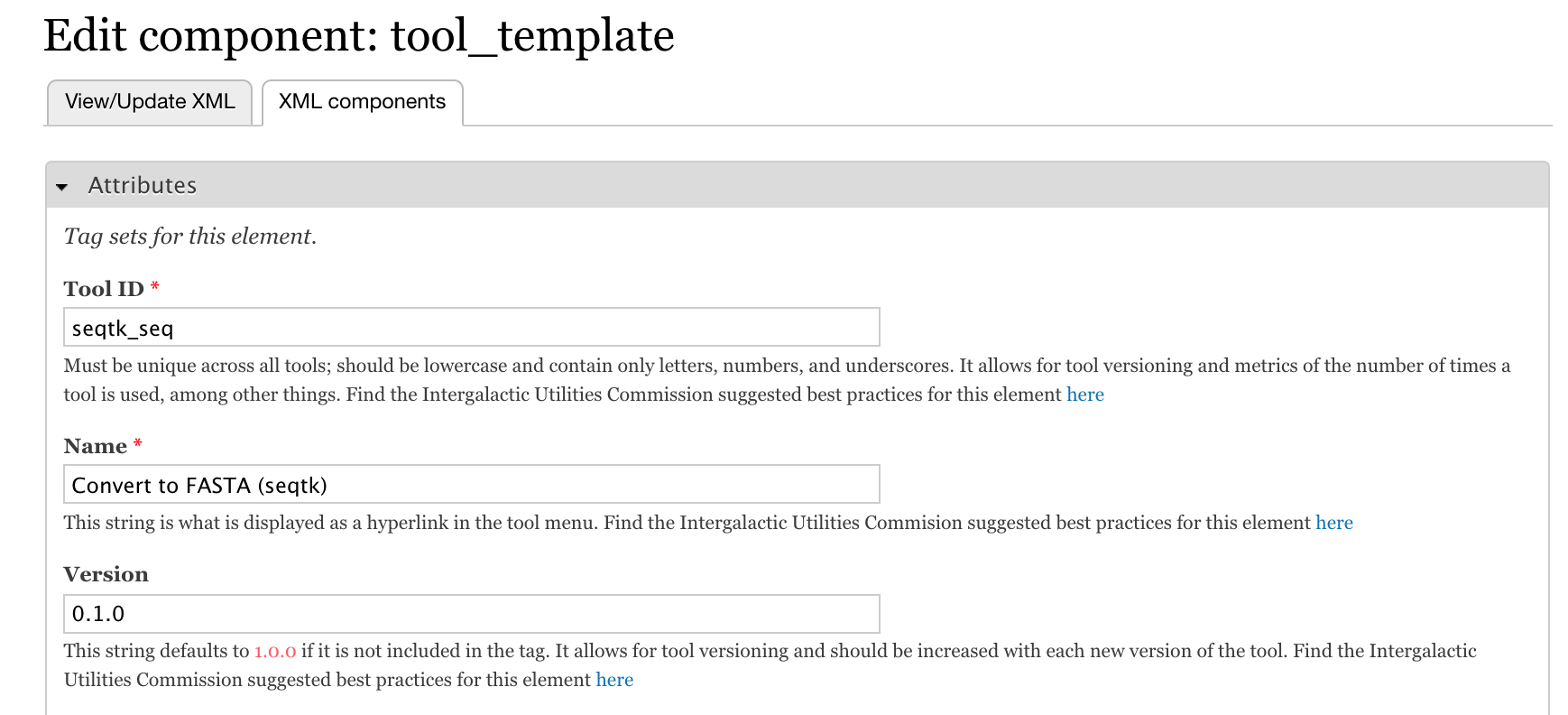
- This will open the edit form for the tool component, through which you can edit the attributes.
- Replace
tool_1withseqtk_seq. - Click Save component
- Replace
Add more components¶
Compared to the seqtk_seq_2.xml, seqtk_seq_1.xml is missing the following components. We are going to add them one by one.
The tool->tests component¶
<tests>
<test>
<param name="input1" value="2.fastq"/>
<output name="output1" file="2.fasta"/>
</test>
</tests>
Add a tests component of the tool->tests component type and drag it to the correct location.
The component tool->test is a subcomponent of the component tool. It needs to be placed under tool and at the same level as other components like tool->requirements, tool->command, tool->inputs, tool->outputs, and tool->help. You can drag a component to arrange its location. All subcomponents needs to be correctly placed under their parent components.
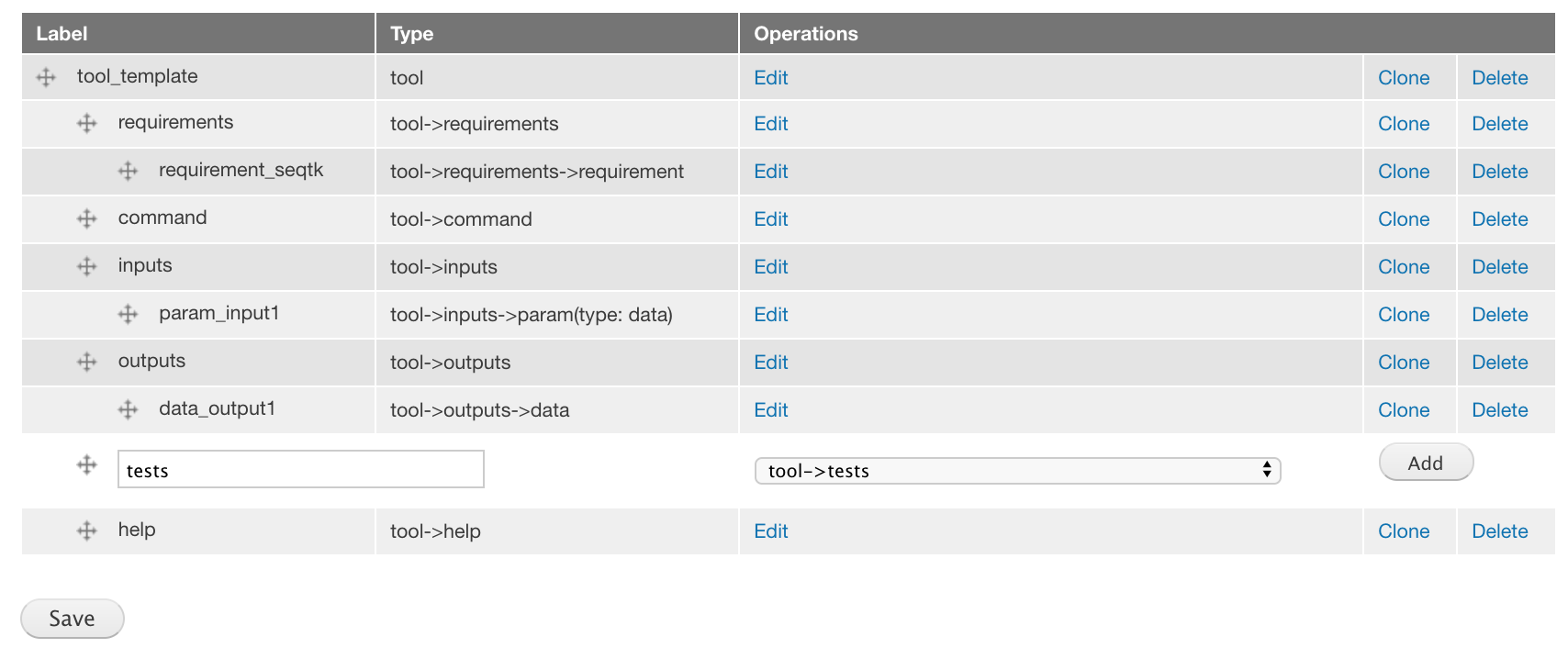
There are no attributes to choose.

Add a test component of the tool->tests->test component type and place it under the tests component.
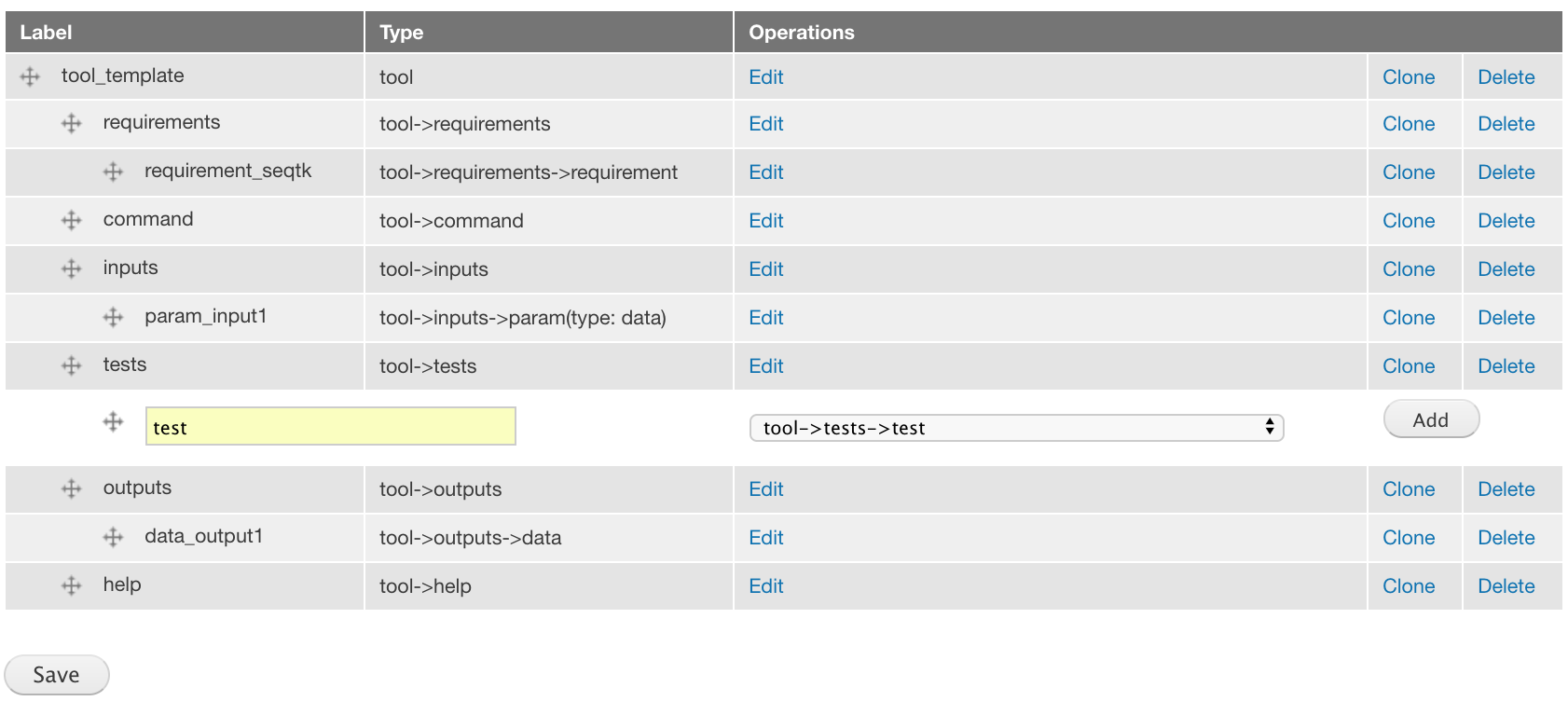
Again, there are no attributes to choose.

Add a tool->tests->test->param component labeled input1.
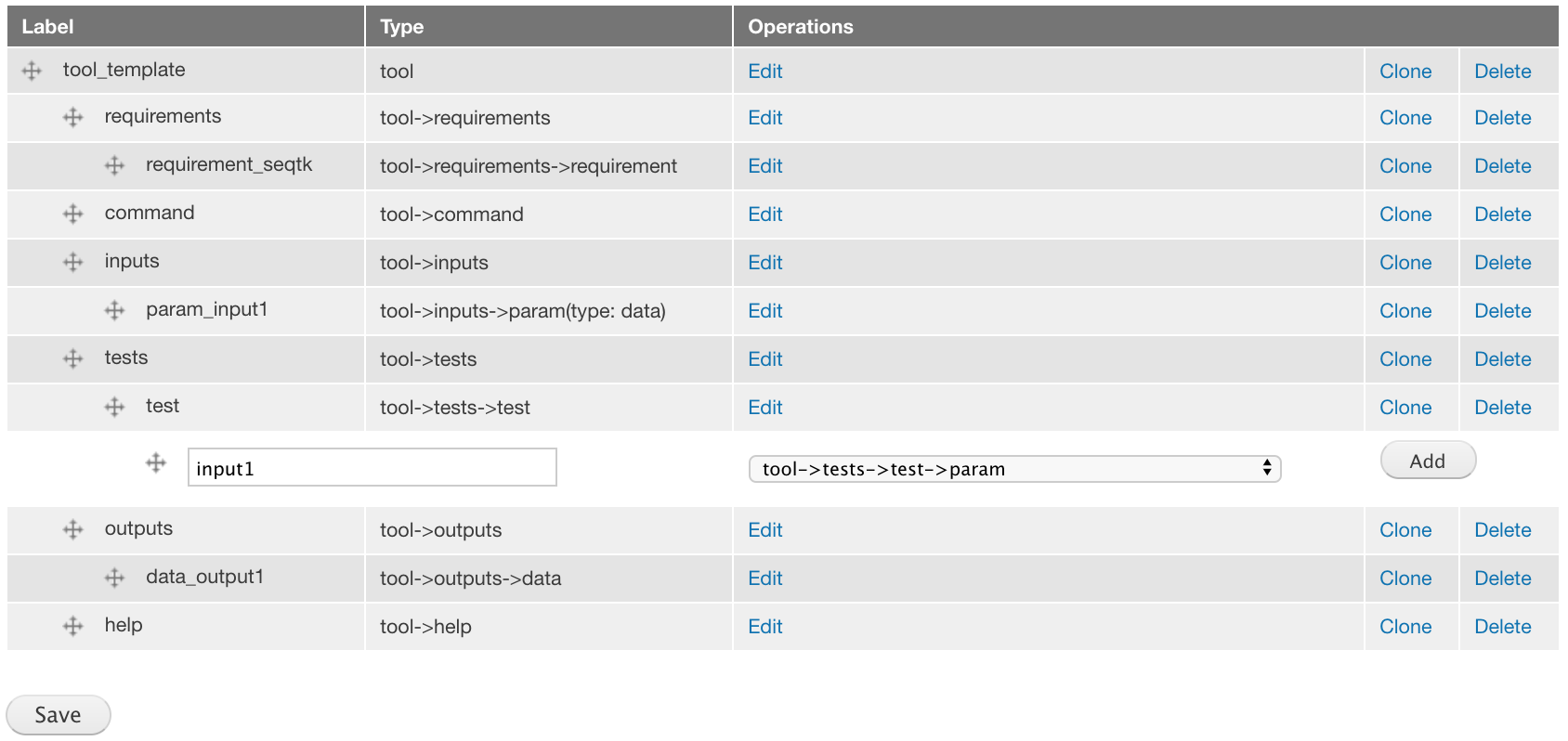
For the attributes, set Name to 2.fastq.
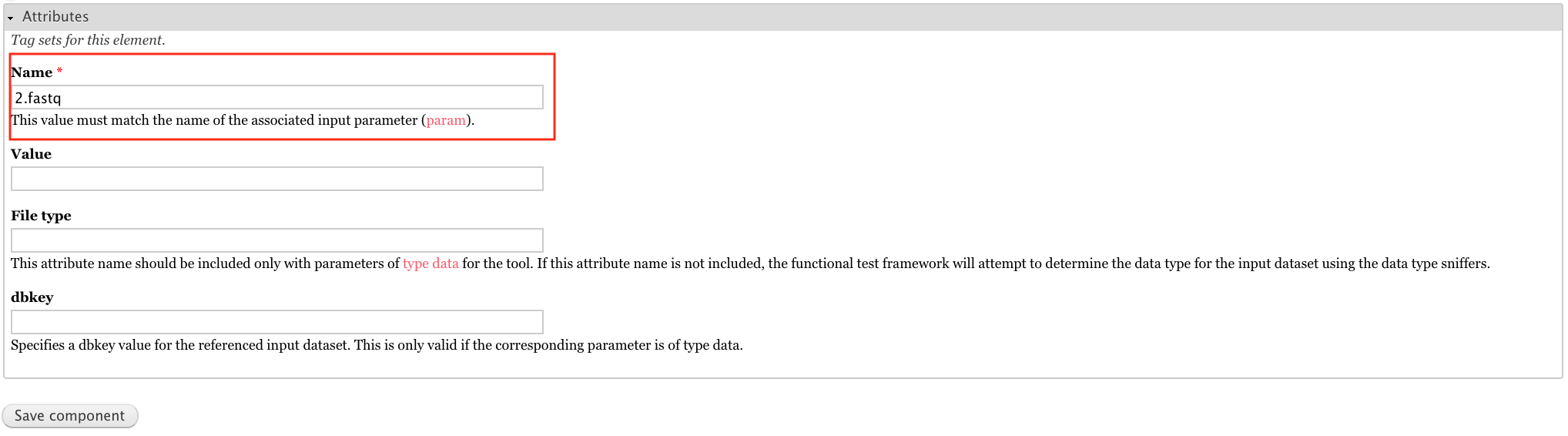
Add a tool->tests->test-output component labeled output1.
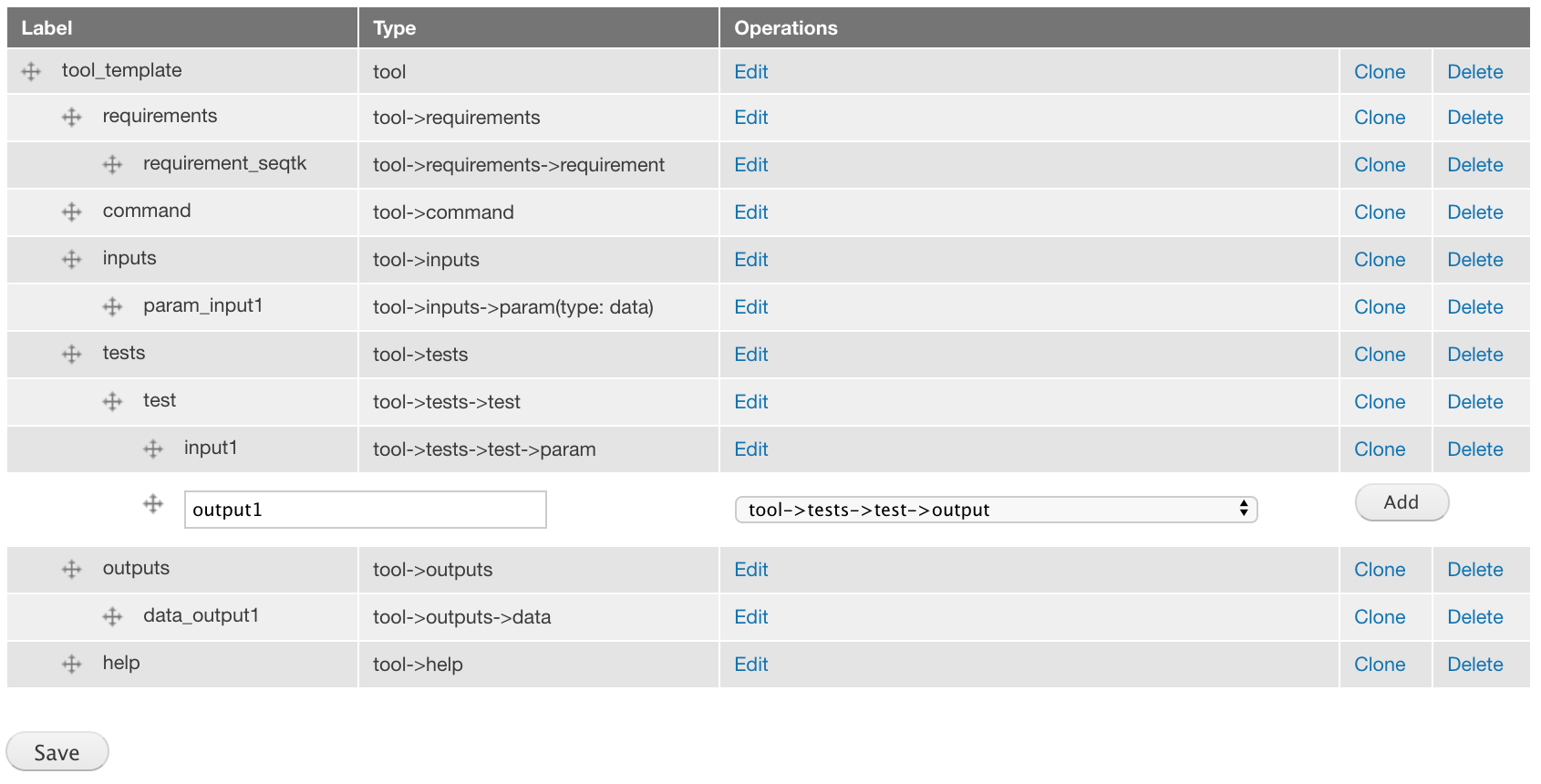
For the attributes, set Name to output1 and File to 2.fasta
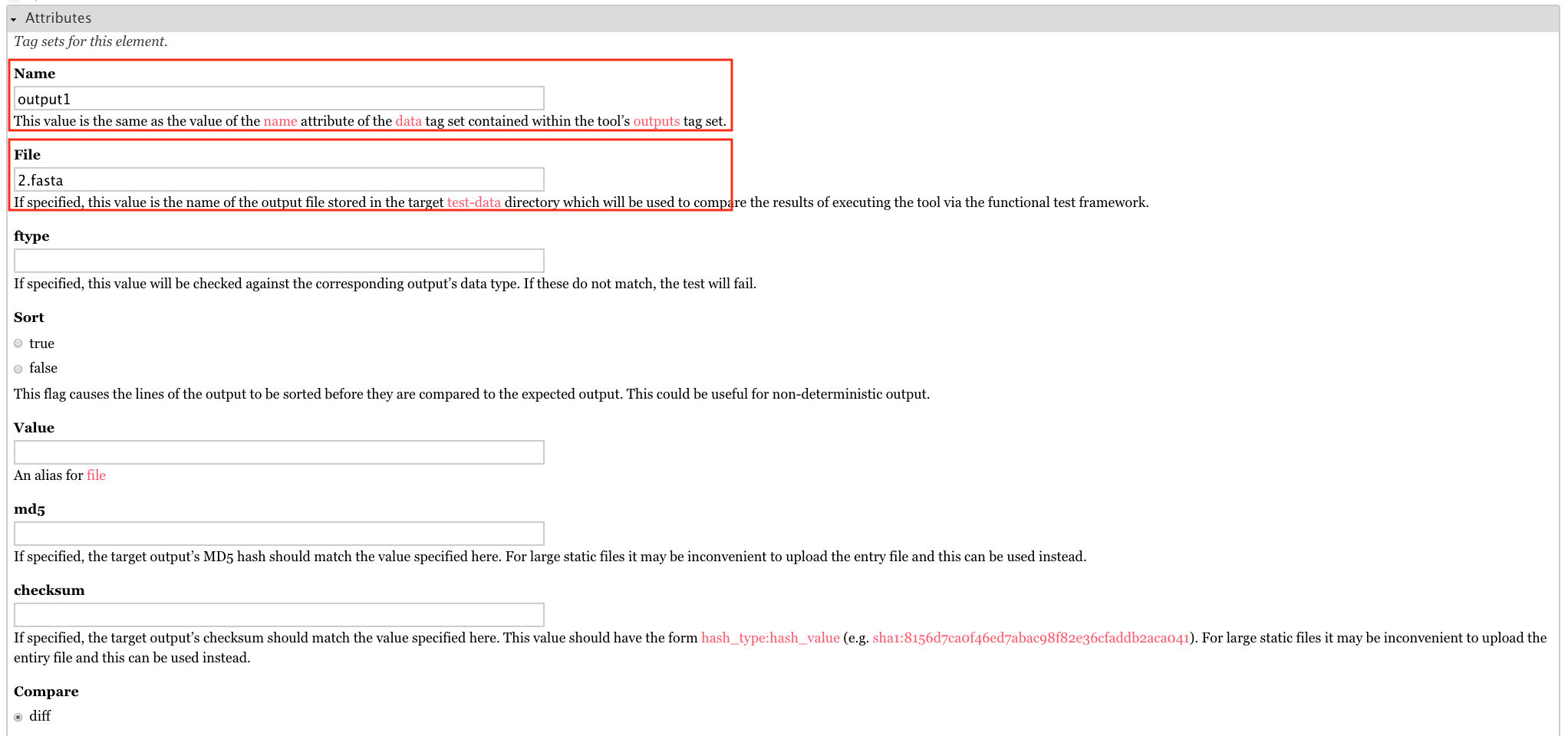
The content in the tool->help component¶
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2^32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
-S strip of white spaces in sequences
The uploaded XML already has a tool->help component. We just need to open the component edit form and fill in the content above.
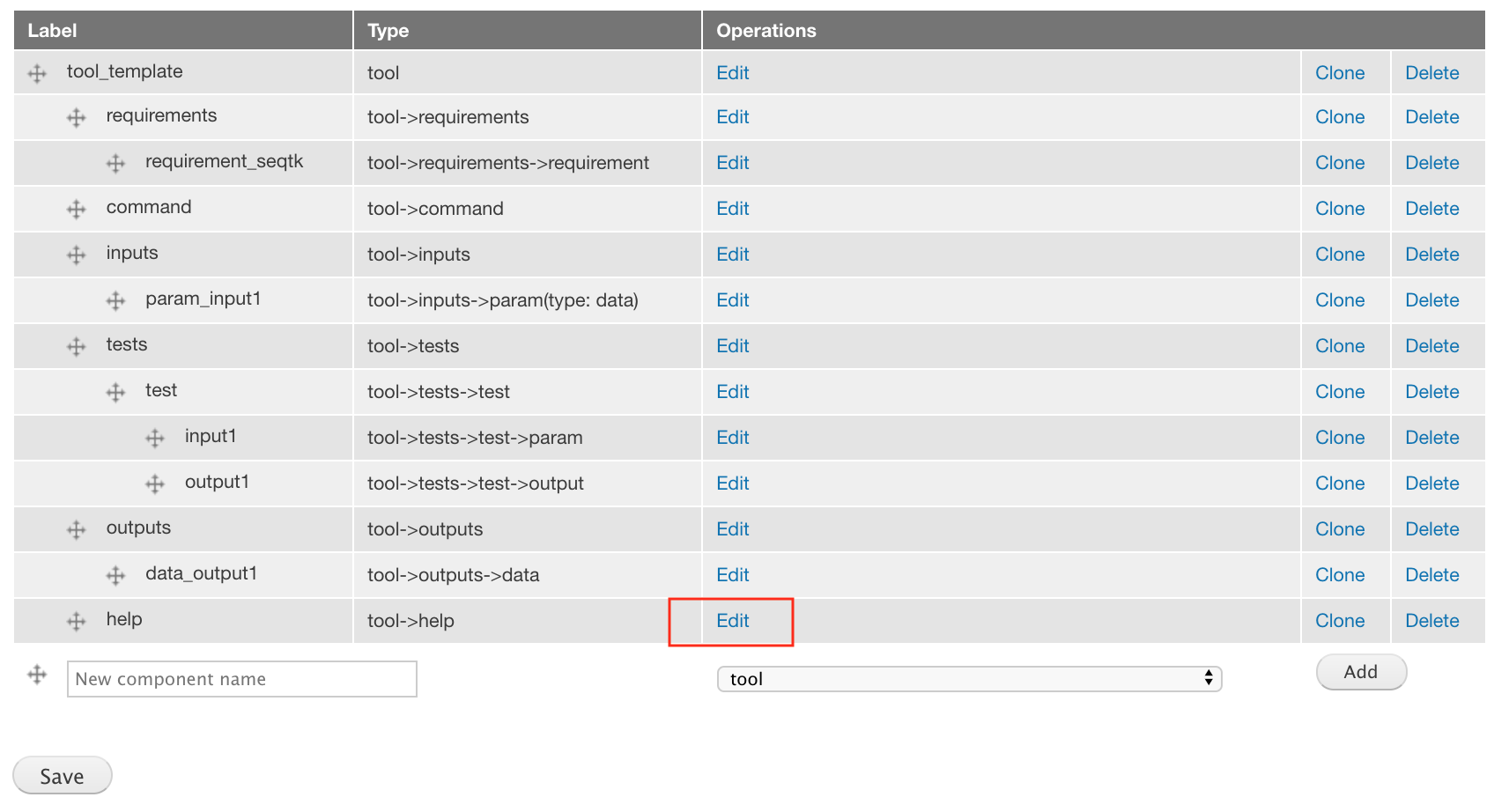
For the attributes, paste the below text into the XML value field.
Usage: seqtk seq [options] <in.fq>|<in.fa>
Options: -q INT mask bases with quality lower than INT [0]
-X INT mask bases with quality higher than INT [255]
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]
-l INT number of residues per line; 0 for 2~32-1 [0]
-Q INT quality shift: ASCII-INT gives base quality [33]
-s INT random seed (effective with -f) [11]
-f FLOAT sample FLOAT fraction of sequences [1]
-M FILE mask regions in BED or name list FILE [null]
-L INT drop sequences with length shorter than INT [0]
-c mask complement region (effective with -M)
-r reverse complement
-A force FASTA output (discard quality)
-C drop comments at the header lines
-N drop sequences containing ambiguous bases
-1 output the 2n-1 reads only
-2 output the 2n reads only
-V shift quality by '(-Q) - 33'
-U convert all bases to uppercases
-S strip of white spaces in sequences
The tool->citations component¶
<citations>
<citation type="bibtex">
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}</citation>
</citations>
Add tool->citations component labeled citations.
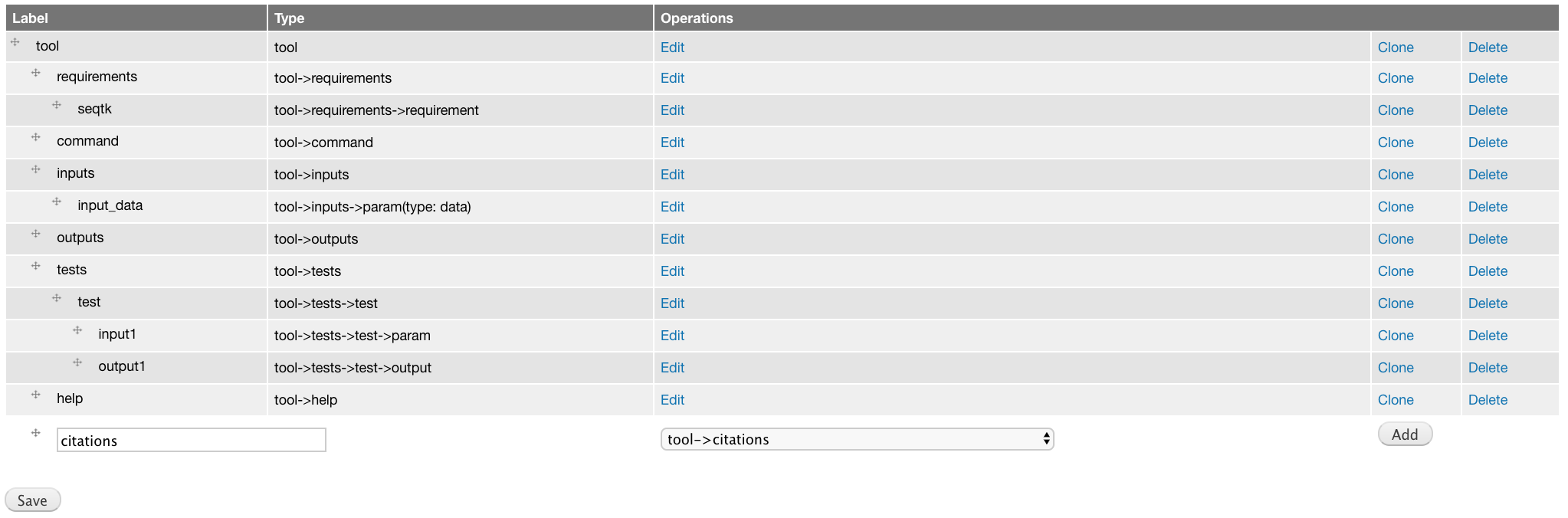
This component does not have attributes.

Add tool->citations->citation component labeled citation githubseqtk.
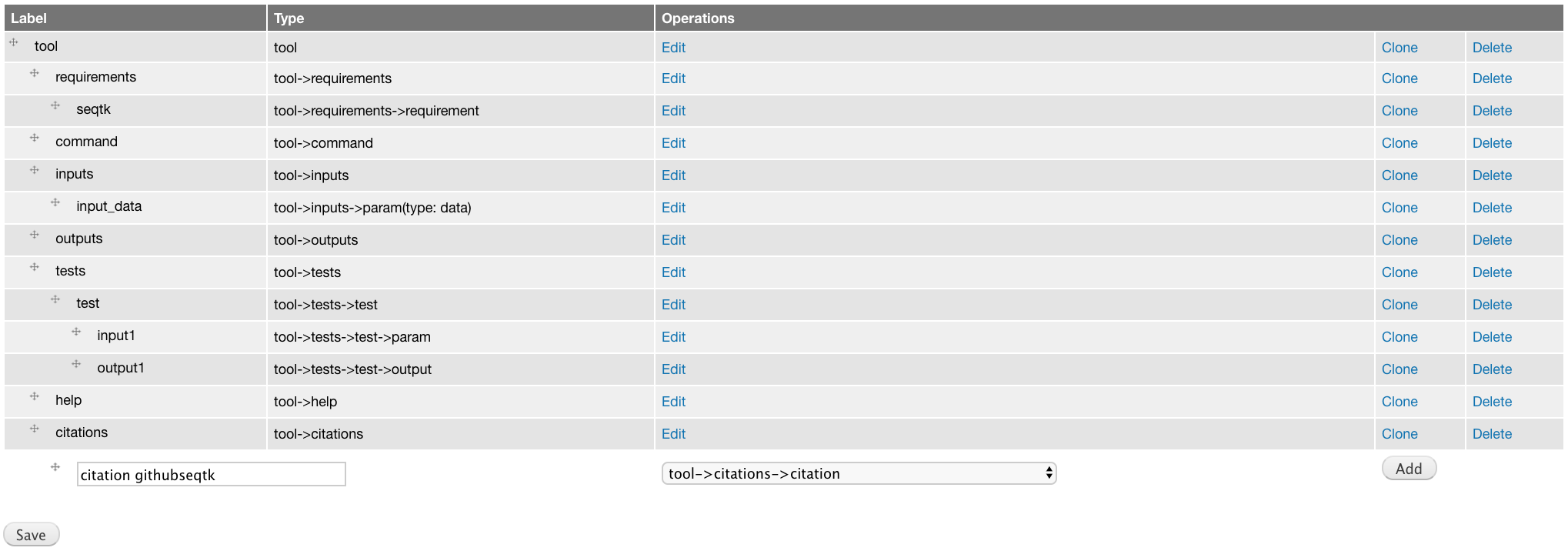
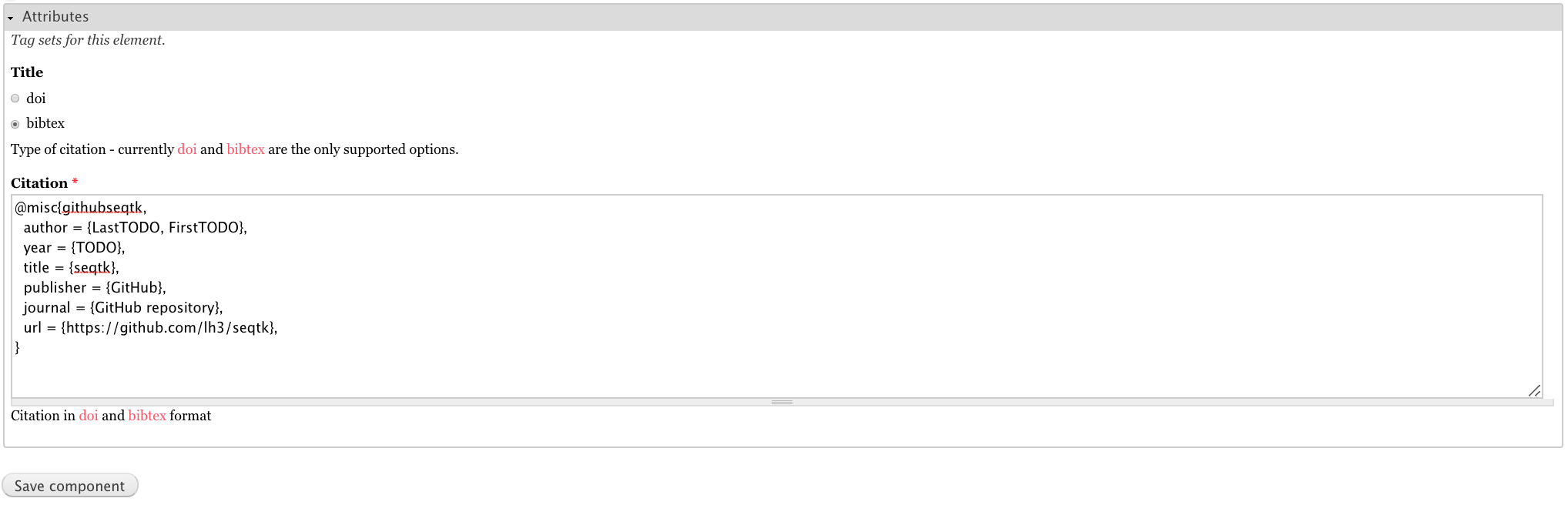
For the attributes, select bibtex for the Title, and paste the below citation in the Citation field.
@misc{githubseqtk,
author = {LastTODO, FirstTODO},
year = {TODO},
title = {seqtk},
publisher = {GitHub},
journal = {GitHub repository},
url = {https://github.com/lh3/seqtk},
}
View the complete XML file¶
To view the complete XML file, you can following the instruction from the From Scratch guide.